
Research project
In our laboratory, we are interested in the study of cell and molecular events associated to virulence, pathogenesis and host-pathogen interactions of C. trachomatis, which is an obligate intracellular bacterium with a high impact in public health globally.
C. trachomatis is the main bacterial cause of sexually transmitted infections and of infectious blindness worldwide. About 70-90% of women with endocervical infections due to C. trachomatis are asymptomatic and thereby do not seek medical treatment and become a reservoir for transmission. The ability of this bacterium to cause recurrent and persistent infections results in serious complications such as pelvic inflammatory disease, ectopic pregnancy, abortion and infertility, mainly in young women. To a lesser extent, genital infections due to C. trachomatis in males may also be asymptomatic and chronic, which also contributes to spreading and sometimes lead to complications like epididymitis and prostatitis.
C. trachomatis propagates through a biphasic cycle involving the elementary body (EB) and the reticulate body (RB). The EB is infectious, environmentally resistant, metabolically dormant, more electron-dense and smaller than the RB. On the other hand, the RB is highly replicative, labile, metabolically active, non-infectious and exhibits a bigger size. After attachment to epithelial cells, the EBs promote their own internalization and get confined in a vacuole termed an “inclusion”. At early stages post-infection, EBs differentiate into RBs, which begin replicating causing the inclusion to expand. Around the mid-cycle, RBs begin to differentiate back into EBs in an asynchronous process. Finally, EBs are released to the extracellular environment (either through cell lysis or extrusion), where other cells can be infected. If during replication Chlamydia encounters stress factors, like those resulting from exposure to INF-gamma (a key cytokine in the immune response) or beta-lactam antibiotics, these bacteria enter in a viable/ non-culturable state which is also known as “persistent form” or “aberrant body”, usually much bigger than the RBs. It’s been demonstrated that Chlamydia may stay in this sort of dormant state for up to 9 months in cell culture, being able to resume normal replication once the stress is removed (Figure 1).
Chlamydial persistence has been linked to the capacity of causing chronic/ recurrent infections both in women and men and with increased antimicrobial resistance, making this a highly relevant phenomenon in chlamydial pathogenesis.
In this context, we are interested in studying the poorly characterized factors involved in chlamydial persistence. In order to identify the chlamydial genes involved in persistence induced by INF-gamma or beta-lactam antibiotics we are using a genomics approach based on chemical mutagenesis and whole genome sequence analysis. Additionally, we are interested in characterizing the proteome of persistent forms in order to identify chlamydial proteins that are differentially expressed during this state.
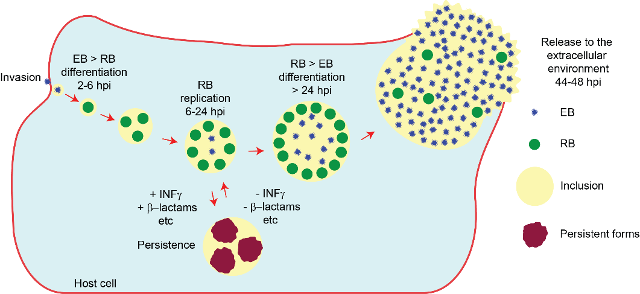
Figure 1. Cartoon representing the life cycle of C. trachomatis serovar LGV-L2. Right after invading epithelial cells, EBs differentiate into RBs which begin replicating by binary fission. Approximately at 20-24 hours post-infection (hpi), RBs begin to asynchronically differentiate back into EBs. Around 44-48 hpi the infectious progeny is released to the extracellular environment. If during replication Chlamydia encounters stressing conditions, like those resulting exposure to gamma-interferon (INFγ) or β-lactam antibiotics, RBs can reversibly differentiate into “persistent forms” or “aberrant bodies”, which sometimes exhibit striking morphological differences like increased size and an irregular shape. In this state, Chlamydia has the capacity of staying viable for long periods of time. Once the stress is removed, Chlamydia resumes normal replication.
Besides persistence, we are also interested in studying other key aspects of C. trachomatis pathogenesis such as nutrient acquisition, immune evasion and Chlamydia-induced reprogramming of the host cell machinery during the infectious cycle.
Publications
Dr. Saka has published numerous scientific papers in prestigious peer-reviewed journals.
For a detailed list, please see: